
Os produtos IVD s├Żo dispositivos e sistemas usados ŌĆŗŌĆŗpara diagnosticar, tratar ou prevenir problemas de sa├║de. Eles devem ser usados ŌĆŗŌĆŗna coleta e exame de amostras biol├│gicas como sangue, saliva ou tecido. As amostras podem ser retiradas de dentro do nariz ou da parte de tr├Īs da garganta, ou de uma veia ou ponta do dedo.
├ē importante observar que os produtos IVD n├Żo s├Żo invasivos. Por exemplo, analisador de imunoensaio de quimioluminesc├¬ncia homog├¬nea Poclight HSCL-5000 Micro .
No entanto, eles ainda s├Żo considerados dispositivos m├®dicos e, portanto, est├Żo sujeitos aos mesmos controles. Dispositivos m├®dicos consistem em qualquer instrumento, aparelho, software ou dispositivo relacionado destinado a ser usado por seres humanos com a finalidade de:
┬Ę Diagnosticar, prevenir, monitorar ou tratar doen├¦as ou les├Ąes.
┬Ę Apoiar ou sustentar a vida.
┬Ę Regula├¦├Żo da concep├¦├Żo.
┬Ę Desinfec├¦├Żo de dispositivos m├®dicos.
┬Ę Fornecimento de informa├¦├Ąes para fins m├®dicos atrav├®s da coleta de amostras biol├│gicas de humanos.
Os fabricantes de Diagn├│sticos In Vitro devem atender aos requisitos regulat├│rios da Uni├Żo Europ├®ia, Reino Unido e Estados Unidos, dependendo do(s) mercado(s) em que desejam penetrar.
Esses requisitos incluem o Regulamento de Diagn├│stico In Vitro (IVDR) (UE) 2017/746 na UE e o Regulamento de Dispositivos M├®dicos do Reino Unido (UK MDR) 2002 no Reino Unido. Nos Estados Unidos, os produtos IVD s├Żo definidos na se├¦├Żo 201(h) da Lei Federal de Alimentos, Medicamentos e Cosm├®ticos (Lei FD&C); esses dispositivos tamb├®m podem estar sujeitos ├Ā se├¦├Żo 351 da Lei do Servi├¦o de Sa├║de P├║blica, bem como ├Ā categoriza├¦├Żo sob as Emendas de Melhoria de Laborat├│rio Cl├Łnico (CLIA) de 1988.
De acordo com o IVDR, um produto IVD ├® definido como ŌĆ£qualquer dispositivo m├®dico que seja um reagente, produto reagente, calibrador, material de controle, kit, instrumento, aparelho, pe├¦a de equipamento, software ou sistema, usado sozinho ou em combina├¦├Żo, destinado pelo fabricante para ser usado in vitro para o exame de amostras, incluindo doa├¦├Ąes de sangue e tecidos, derivadas do corpo humanoŌĆ”ŌĆØ
Especialistas estimam que aproximadamente 70% de todas as decis├Ąes cl├Łnicas s├Żo feitas usando produtos IVD.
Testes de gravidez, testes COVID-19 e testes de HIV s├Żo exemplos de produtos IVD. Outros exemplos de dispositivos Poclight HSCL-5000 Micro CLIA Analyzer IVD incluem:
┬Ę Diagn├│stico de c├óncer
┬Ę Diagn├│stico da tireoide
┬Ę Diagn├│stico de inflama├¦├Żo
┬Ę Imunoensaios
┬Ę ...
Os recept├Īculos fabricados para amostras m├®dicas tamb├®m s├Żo produtos IVD. De acordo com a Organiza├¦├Żo Mundial da Sa├║de, mais de 40.000 produtos est├Żo dispon├Łveis para testes de IVD hoje. Estes podem variar de testes laboratoriais tradicionais a testes no local de atendimento.
Nos Estados Unidos, a FDA classifica todos os dispositivos m├®dicosŌĆöincluindo produtos IVDŌĆöcomo Classe I, Classe II ou Classe III. A classifica├¦├Żo do dispositivo varia de acordo com o risco envolvido e o n├Łvel de controle regulat├│rio necess├Īrio para garantir a seguran├¦a do produto. Assim, a classifica├¦├Żo IVD determinar├Ī o processo de pr├®-comercializa├¦├Żo que o fabricante precisa seguir para levar seu produto ao mercado.
┬Ę Os produtos Classe I s├Żo considerados de risco baixo a moderado e requerem controles gerais.
┬Ę Os produtos Classe II s├Żo considerados de risco moderado a alto e requerem controles gerais e controles especiais.
┬Ę Os produtos Classe III s├Żo considerados de alto risco e requerem controles gerais e Aprova├¦├Żo Pr├®-Mercado (PMA).
├ē importante observar que, ├Ā medida que a classe do dispositivo aumenta, os controles regulat├│rios tamb├®m aumentam. Os dispositivos IVR Classe I est├Żo sujeitos ao menor controle regulat├│rio, enquanto os dispositivos IVR Classe III apresentam os requisitos mais rigorosos. As empresas de ci├¬ncias da vida que desejam levar seus produtos de diagn├│stico in vitro ao mercado nos Estados Unidos podem consultar o recurso ŌĆ£Classifique seu dispositivo m├®dicoŌĆØ da FDA.
Procurando garantir a conformidade regulat├│ria na Uni├Żo Europeia? O IVDR foi atualizado em 2021 para incluir melhorias como:
┬Ę Obriga├¦├Ąes claras para fabricantes, importadores e distribuidores envolvendo rastreabilidade de dispositivos, registro e verifica├¦├Żo de rotulagem adequada. Isso inclui um sistema de rastreabilidade baseado em um identificador ├║nico de dispositivo (UDI).
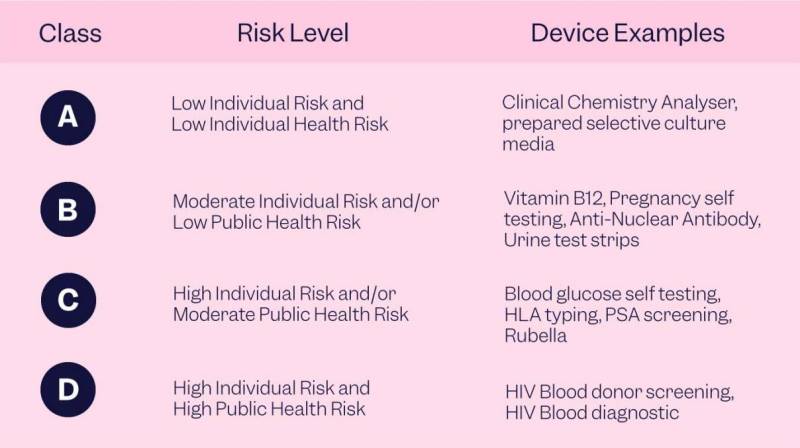
┬Ę Introdu├¦├Żo de uma classifica├¦├Żo composta por quatro classes de risco. Os dispositivos Classe A apresentam baixo risco individual e ├Ā sa├║de p├║blica, os dispositivos Classe B apresentam risco individual moderado e/ou baixo risco ├Ā sa├║de p├║blica, os dispositivos Classe C apresentam alto risco individual e/ou risco moderado ├Ā sa├║de p├║blica e os dispositivos Classe D apresentam alto risco individual e alto risco ├Ā sa├║de p├║blica.
┬Ę Controles mais r├Łgidos para dispositivos IVD de alto risco.
┬Ę Garantia de supervis├Żo dos organismos notificados (ou organismos independentes de avalia├¦├Żo da conformidade de terceiros) atrav├®s do refor├¦o de um conjunto espec├Łfico de crit├®rios.
┬Ę Maior transpar├¬ncia com uma base de dados abrangente sobre dispositivos m├®dicos na UE (chamada EUDAMED).
┬Ę Evid├¬ncias cl├Łnicas refor├¦adas e regulamentos de avalia├¦├Żo de desempenho, com maior coordena├¦├Żo entre os pa├Łses da UE ŌĆō especialmente no que diz respeito ├Ā fiscaliza├¦├Żo do mercado.
┬Ę Requisitos de vigil├óncia p├│s-comercializa├¦├Żo mais fortes do fabricante.
┬Ę Requisitos espec├Łficos para ŌĆ£dispositivos internosŌĆØ ou produtos IVD fabricados e usados ŌĆŗŌĆŗna mesma institui├¦├Żo.
Colocar um produto IVD no mercado com seguran├¦a e efic├Īcia ├® fundamental ŌĆō e entender os regulamentos em vigor ├® o primeiro passo ideal.
Os produtos IVD s├Żo uma linha cr├Łtica de defesa contra v├Īrias condi├¦├Ąes de sa├║de. A seguran├¦a e a qualidade do Diagn├│stico In Vitro s├Żo essenciais, e ├® exatamente por isso que a conformidade regulat├│ria do fabricante ├® t├Żo importante. A Poclight ├® certificada pela CE e ISO e possui uma gama completa de itens de reagentes para tornar os testes sem preocupa├¦├Ąes.






 Rede IPv6 suportada | XML | Sitemap | Blog | pol├Łtica de Privacidade
Rede IPv6 suportada | XML | Sitemap | Blog | pol├Łtica de Privacidade
Deixe um recado
Digitalizar para WeChat :


 English
English fran├¦ais
fran├¦ais čĆčāčüčüą║ąĖą╣
čĆčāčüčüą║ąĖą╣ espa├▒ol
español português
portugu├¬s ž¦┘äž╣ž▒ž©┘Ŗž®
ž¦┘äž╣ž▒ž©┘Ŗž® µŚźµ£¼Ķ¬×
µŚźµ£¼Ķ¬× T├╝rk├¦e
T├╝rk├¦e Óż╣Óż┐ÓżéÓż”ÓźĆ
Óż╣Óż┐ÓżéÓż”ÓźĆ Indonesia
Indonesia